
You are using an out of date browser. It may not display this or other websites correctly.
You should upgrade or use an alternative browser.
You should upgrade or use an alternative browser.
Coronavirus Thread: Worldwide Pandemic
- Thread starter 88m3
- Start date
More options
Who Replied?newworldafro
DeeperThanRapBiggerThanHH
Lab grown Frankensteined/Schwarzaneggered lil fukker.
Chinese doctors say coronavirus ‘like a combination of SARS and AIDS’
Chinese doctors say coronavirus ‘like a combination of SARS and AIDS’
Lab grown Frankensteined/Schwarzaneggered lil fukker.
Chinese doctors say coronavirus ‘like a combination of SARS and AIDS’
An airbourne version of AIDS?

newworldafro
DeeperThanRapBiggerThanHH
An airbourne version of AIDS?
House In the enVironment.
Built-a-Thot?
No this is Build-a-Virus.
This is close to being a normal community virus which is going to be detrimental. What do you do as children to older parents/grandparents? Do we take precautions by staying away from them eventually
Last edited:
Lab grown Frankensteined/Schwarzaneggered lil fukker.
Chinese doctors say coronavirus ‘like a combination of SARS and AIDS’
An airbourne version of AIDS?
That was a super sensationalized title compared to what was actually in the story. The quote from the doctor I don't think was meant to conjure up quite those images.
This is close to being a normal community virus which is goinf to be detrimental. What do you do as children to oldee parents/grandparents? Do we take precautions by staying away from them eventually
I've been wondering about this too. Hard to know who to spend extra time to help and who to keep away from everyone else including yourself.
Man I’m kind of afraid of going to the gym now. Who knows who got what? I always wipe down treadmills, weight sets etc but you know shyt is brewing with all those sweaty ass people
And I'd take this virus rather than be poisoned by money on the brain
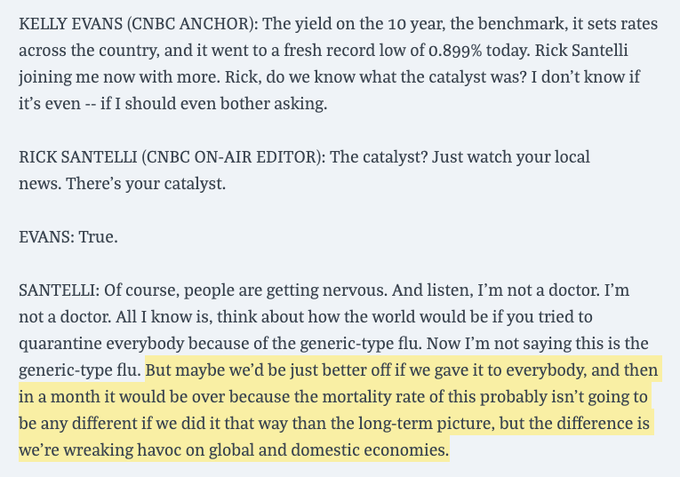
I saw another one today that gave me the same thought.
"We're getting to the point where the reaction to the virus may be worse than the virus itself," said Nicholas Calio of Airlines for America.
Aviation industry feeling 'gut punch' from coronavirus, executives say - CNNPolitics
Yeah, people not flying as much for a few weeks is worse than thousands of deaths.

One day we'll be grateful for Coronavirus ending Trump's presidency
I think that's way too big an assumption for now. If the people who follow the markets really really believed that the drops would have been even bigger already. We still don't know exactly how this is all going to play out.The stock market is in a free-for-all drop and about to wipe out all the gains of the last 4 years. If this keeps up another week, a whole bunch of job losses will occur.
The Coronavirus really bodied the Trump presidency.
newworldafro
DeeperThanRapBiggerThanHH
That was a super sensationalized title compared to what was actually in the story. The quote from the doctor I don't think was meant to conjure up quite those images.
Breh!!! This virus having HIV/AIDs components was discussed over a month ago when the New Delhi, India research institute said this exact thing.
Alt/Indy media sites began reporting the results of the study and original articles. Per unusual, the MsM said it was conspiracy theory.
Within days the Indian government, under apparent pressure, forced the research lab to retract the HIV/AIDS part of the study....although the researchers say they voluntarily withdrew that part...................but now Chinese scientist saying it has HIV/AIDS components, and it's being treated with HIV/AIDS retrovirals.
"What are the odds?" - A timeline of facts linking COVID-19, HIV, & Wuhan's secret bio-lab

COVID-19 has a unique sequence about 1,378 nucleotide base pairs long that is not found in related coronaviruses. They claimed to identify genetic similarities in this unique material between COVID-19 and HIV-1.
Specifically, they isolated 4 short genetic sequences in key protein structures (the receptor binding domain, or RBD).
Two of the sequences were perfect matches (albeit, short), and two of the sequences were matched but each with an additional string of non-matching material appearing in the middle of the sequence.
The paper was criticized and numerous attempts have been made to debunk it. After the criticism, the authors voluntarily withdrew it, intending to revise it based on comments made about their technical approach and conclusions.
One key debunking attempt claims this:
The same sequences are found in a variant called BetaCoV/bat/Yunnan/RaTG13/2013, which had been found “in the wild” in bats.
This is an attempt to prove that it was not engineered, but mutated naturally in the wild.
But there’s a problem…
This strain was only known by and studied at the Wuhan Virology Institute, and although they claim it was discovered in 2013, it wasn’t published or shared with the scientific community until immediately after the Indian paper, on January 27, 2020.

The RatG13 strain publication and the HIV research paper from 2008 share an author.
I discovered this on my own by comparing the two papers and then quickly realized this scientist’s contact information was the information that ZeroHedge was suspended from Twitter for sharing.
Their article identifies this author in question including some contact information from the Wuhan Virology Institute web site.
COVID-19 has a unique sequence about 1,378 nucleotide base pairs long that is not found in related coronaviruses. They claimed to identify genetic similarities in this unique material between COVID-19 and HIV-1.
Specifically, they isolated 4 short genetic sequences in key protein structures (the receptor binding domain, or RBD).
Two of the sequences were perfect matches (albeit, short), and two of the sequences were matched but each with an additional string of non-matching material appearing in the middle of the sequence.
The paper was criticized and numerous attempts have been made to debunk it. After the criticism, the authors voluntarily withdrew it, intending to revise it based on comments made about their technical approach and conclusions.
One key debunking attempt claims this:
The same sequences are found in a variant called BetaCoV/bat/Yunnan/RaTG13/2013, which had been found “in the wild” in bats.
This is an attempt to prove that it was not engineered, but mutated naturally in the wild.
But there’s a problem…
This strain was only known by and studied at the Wuhan Virology Institute, and although they claim it was discovered in 2013, it wasn’t published or shared with the scientific community until immediately after the Indian paper, on January 27, 2020.
The RatG13 strain publication and the HIV research paper from 2008 share an author.
I discovered this on my own by comparing the two papers and then quickly realized this scientist’s contact information was the information that ZeroHedge was suspended from Twitter for sharing.
Their article identifies this author in question including some contact information from the Wuhan Virology Institute web site.
Last edited:
It was retracted because it was bullshyt. The paper hadn't been reviewed by anyone and within hours more careful scientists were poking a thousand holes in the claim. Some easy examples of that are in the Twitter thread here:Breh!!! This virus having HIV/AIDs components was discussed over a month ago when the New Delhi, India research institute said this exact thing.
Alt/Indy media sites began reporting the results of the study and original articles. Per unusual, the MsM said it was conspiracy theory.
Within days the Indian government, under apparent pressure, forced the research lab to retract the HIV/AIDS part of the study....although the researchers say they voluntarily withdrew that part...................but now Chinese scientist saying it has HIV/AIDS components, and it's being treated with HIV/AIDS retrovirals.
You don't have to follow the mainstream media on this if you don't want to. There are tens of thousands of microbiologists and infectious disease specialists all over the world who have the same access to that article that you do. I've been part of that world before - trust me, they aren't a bunch of unthinking establishment lackeys and they aren't obeying instructions from a hive mind. If there is any legitimacy to the paper or any real connection between COVID-19 and HIV, you will see plenty of virologists talking about it no matter what the mainstream media says.
Here's a more formal comment declaring no contribution from HIV-1 in COVID-19. It's a good read - the conspiracy falls apart on at least different levels - the "similarities" aren't actually very similar, those particular segments are much closer to other nearby viruses from the same virus family, and the insertions don't appear to give the virus any particular advantage.
https://www.tandfonline.com/doi/full/10.1080/22221751.2020.1727299
HIV-1 did not contribute to the 2019-nCoV genome
Chuan Xiao,Xiaojun Li
 ,Shuying Liu,Yongming Sang,Shou-Jiang Gao &Feng Gao
,Shuying Liu,Yongming Sang,Shou-Jiang Gao &Feng Gao
Pages 378-381 | Received 04 Feb 2020, Accepted 04 Feb 2020, Published online: 14 Feb 2020

When a new pathogen that causes a global epidemic in humans, one key question is where it comes from. This is especially important for a zoonotic infectious disease that jumps from animals to humans. Knowing the origin of such a pathogen is critical to develop means to block further transmission and to develop vaccines. Discovery of the origin of a newly human pathogen is a sophisticated process that requires extensive and vigorous scientific validations and generally takes many years, such as the cases for HIV-1 [1], SARS [2] and MERS [3]. Unfortunately, before the natural sources of new pathogens are clearly defined, conspiracy theories that the new pathogens are man-made often surface as the source. However, in all cases, such theories have been debunked in history.
Infection from an emerging pathogenic coronavirus was first reported in December 2019 in China. It has now affected over 42,000 people and caused over 1,000 deaths in 25 countries (https://2019ncov.Chinacdc.Cn/2019-Ncov). The complete genome of this new virus was quickly sequenced and made public on January 12, only about 2 weeks after the disease was first observed [4]. It was named as 2019-nCoV the following day by the World Health Organization (WHO). Phylogenetic analysis shows that 2019-nCoV is a new member of coronaviruses that infect humans. It is genetically homogenous but distinct from coronaviruses that cause SARS and MERS [5,6]. However, it shares a high level of genetic similarity (96.3%) with a bat coronavirus RaTG13 which was obtained from bat in Yunnan in 2013, suggesting that RaTG13-like viruses are most likely the reservoir, but not the immediate sources of the current 2019-nCoV viruses [7].
Lack of the definite origin of 2019-nCoV has led to speculation that 2019-nCoV might be derived from genetic manipulation or even for the purpose of use as a bioweapon. This notion has been fully debunked in the media. A recent informally presented report, however, showed that 2019-nCoV had four insertions in the spike glycoprotein gene that is critical for the virus to enter the target cells when compared to other coronaviruses [8]. It was claimed that these inserts were either identical or similar to the motifs in the highly variable (V) regions (V1, V4 and V5) in the envelope glycoprotein or in the Gag protein of some unique HIV-1 strains from three different countries (Thailand, Kenya and India). Together with the structure modelling analysis, the authors speculated that these motif insertions sharing similarity with HIV-1 proteins could provide an enhanced affinity towards host cell receptors and increase the range of host cells of 2019-nCoV. This study implies that 2019-nCoV might be generated by gaining gene fragments from the HIV-1 genome.
Current report conducted careful examination of the sequences of 2019-nCoV, other CoV viruses and HIV-1 as well as GenBank database. Our results demonstrated no evidence that the sequences of these four inserts are HIV-1 specific or the 2019-nCoV viruses obtain these insertions from HIV-1. First, the results of blast search of these motifs against GenBank shows that the top 100 identical or highly homologous hits are all from host genes of mammalian, insects, bacterial and others. There are only a few hits on coronaviruses, but none of them are HIV-1 related. Blast against viral sequence database also showed these insertion sequences widely exist in all kinds of viruses from bacteriophage, influenza, to giant eukaryotic viruses (https://www.hiv.lanl.gov/components/sequence/HIV/search/search.html) yielded similar results. Sequences that completely match the insertion 3 and 4 sequences were not found in any HIV-1 sequences. This clearly shows that these insertioin sequences are widely present in living organisms including viruses, but not HIV-1 specific. All these regions in HIV-1 envelope glycoprotein are highly variable with many large insertions and deletions, indicating that they are not essential for biological functions of HIV-1 envelope glycoprotein. The detection of completely matched sequences of 1 and 2 insertions in only a few HIV-1 strains demonstrated that four insertions are very rare or not present among tens of thousands of natural HIV-1 sequences. This also explains why four insertion homolog sequences could only be independently found in different HIV-1 genomes [8]. Because of their poor identities to and rareness in the HIV-1 sequences, HIV-1 could not be the source for those insertion sequences in the 2019-nCoV genome.
Second, these insertions are present not only in 2019-nCoV viruses but also in three betaCoV sequences from bats: two (ZC45 and ZXC21) from Zhejiang deposited in GenBank in 2018 and RaTG13 from Yunnan obtained in 2013 [8]. The RaTG13 is much more similar to 2019-nCoV than both ZC45 and ZXC21 (Figure 1A). The similarity of the spike protein between RaTG13 and 2019-nCoV is 97.7%. In the RaTG13 genome, two inserts are identical (HKNNKS and RSYLTPGDSSSG) to those in 2019-nCoV, one has one T → I substitution (TNGIKR), and the fourth one misses the C-terminal 4 amino acids (QTNS----) (Figure 1B). ZC45 and ZXC21 are more divergent from 2019-nCoV than RaTG13, but both also contain similar insertions at three insertion sites, except insertion 4 (Figure 1B). Furthermore, many other CoV viruses have similar insertions but with different sequences at the insertion 1 position. These results clearly show that three out of four of these inserts naturally exist in three bat CoV viruses before 2019-nCoV was identified. This undoubtedly refutes the possibility that 2019-nCoV is generated through obtaining gene fragments from the HIV-1 genome. Instead, it is much more likely that 2019-nCoV originated from RaTG13-like CoV viruses.
Figure 1. Sequence and structure analysis of 2019-nCoV and bat coronaviruses. (A) Phylogenetic tree analysis of the spike gene sequences. (B) Sequence alignment of suspected insertion sites between the 2019-nCoV and bat coronavirus sequences. The deletions in the alignment are shown as dashes. The numbers of insertions are indicated at the top of the alignment. (C) Structure comparison of the four insertions in the CoV spike protein and HIV-1 gp120. 2019-nCoV structure was modelled using I-TASSER server with default parameters. Only relevant domains with residues 1 to 708 (exclude residues from 305 to 603) were presented as ribbon diagram. The four insertions were labelled and coloured in red, blue, green and magenta, respectively. HIV-1 gp120 structure (PDB 1GC1) is presented as ribbon diagram. V4, V5, V1/V2 and LE loops were labelled and coloured in red, blue, green, and black, respectively.

Display full size
Third, insertions 1 and 2 in 2019-nCoV have 6-AA motifs identical to those in V4 and V5 of certain HIV-1 gp120 isolates, which are structurally close to each other but separated by a LE loop (Figure 1C) [9]. However, insertion 3 located between insertions 1 and 2 in 2019-nCoV has sequences similar (with deletions) to those in the V1 region of HIV-1 gp120. V1 is far away from V4 and V5 on the opposite side of gp120, which should not interact with V4/V5 in gp120 (Figure 1C) but is now inserted between V4 and V5 in the modelled the 2019-nCoV spike protein structure [10]. Insertion 4 was found in Gag protein of HIV-1 that is not associated with viral entry. This insertion is located too far to be considered to form the same structural unit with the other three insertions in the 2019-nCoV spike protein (Figure 1C). We do not see any selection benefit or rationale for 2019-nCoV to obtain and mix structurally unrelated parts of HIV-1 to generate a unique structure for its enhanced receptor binding as indicated by the authors [8].
How the three bat CoV viruses obtain those inserts remains unknown. For any virus to obtain additional insert sequences from other organisms, it requires that it has direct interactions with other organisms, most likely through homologous or non-homologous recombination [11]. For bat CoV viruses to gain the gene fragments from HIV-1, it will require both viruses to co-infect the same cells. Because the host cells for bat CoV viruses and HIV-1 are different, the chance for both to exchange genetic materials is negligible. On the contrary, these motifs are widely present in various mammalian cells and so it will be more likely for bat CoV viruses to gain those motifs from the genomes of their infected cells if recombination indeed occurs. However, extensive studies of more CoV viruses in wild and domestic animals are warranted to address this question.
Identification of the origins of these inserted sequences in three bat CoV viruses and the new epidemic 2019-nCoV strain will be important for us to understand how CoV viruses jump from animals to humans and adapt in the latter. Current data showed that RaTG13 is most closely related to 2019-nCoV [7]. However, the genetic difference between them is too high for RaTG13 to serve as the immediate ancestor of 2019-nCoV. Other viruses that are more closely related to 2019-nCoV in intermediate animals like civet for SARS and camel for MERS [3,12] are remained to be identified. More studies are necessary to identify the real source of 2019-nCoV. This may take a long time to identify the origin of 2019-nCoV by screening a large number of wild and domestic animals. In any case, reducing or eliminating direct contacts with wild animals will be critical to control the new epidemic infection diseases in the future.
The advances in bioinformatics analysis tools are widely used to easily and rapidly analyse newly obtained sequences. However, great care is required for comprehensive and thorough analysis to fully understand the real biological implications of the new genomic information. Biased, partial and incorrect analysis can dangerously lead to conclusions that fuel conspiracies and harm the process of true scientific discoveries and the effort to control the damage to public health.
Chuan Xiao,Xiaojun Li
Pages 378-381 | Received 04 Feb 2020, Accepted 04 Feb 2020, Published online: 14 Feb 2020
When a new pathogen that causes a global epidemic in humans, one key question is where it comes from. This is especially important for a zoonotic infectious disease that jumps from animals to humans. Knowing the origin of such a pathogen is critical to develop means to block further transmission and to develop vaccines. Discovery of the origin of a newly human pathogen is a sophisticated process that requires extensive and vigorous scientific validations and generally takes many years, such as the cases for HIV-1 [1], SARS [2] and MERS [3]. Unfortunately, before the natural sources of new pathogens are clearly defined, conspiracy theories that the new pathogens are man-made often surface as the source. However, in all cases, such theories have been debunked in history.
Infection from an emerging pathogenic coronavirus was first reported in December 2019 in China. It has now affected over 42,000 people and caused over 1,000 deaths in 25 countries (https://2019ncov.Chinacdc.Cn/2019-Ncov). The complete genome of this new virus was quickly sequenced and made public on January 12, only about 2 weeks after the disease was first observed [4]. It was named as 2019-nCoV the following day by the World Health Organization (WHO). Phylogenetic analysis shows that 2019-nCoV is a new member of coronaviruses that infect humans. It is genetically homogenous but distinct from coronaviruses that cause SARS and MERS [5,6]. However, it shares a high level of genetic similarity (96.3%) with a bat coronavirus RaTG13 which was obtained from bat in Yunnan in 2013, suggesting that RaTG13-like viruses are most likely the reservoir, but not the immediate sources of the current 2019-nCoV viruses [7].
Lack of the definite origin of 2019-nCoV has led to speculation that 2019-nCoV might be derived from genetic manipulation or even for the purpose of use as a bioweapon. This notion has been fully debunked in the media. A recent informally presented report, however, showed that 2019-nCoV had four insertions in the spike glycoprotein gene that is critical for the virus to enter the target cells when compared to other coronaviruses [8]. It was claimed that these inserts were either identical or similar to the motifs in the highly variable (V) regions (V1, V4 and V5) in the envelope glycoprotein or in the Gag protein of some unique HIV-1 strains from three different countries (Thailand, Kenya and India). Together with the structure modelling analysis, the authors speculated that these motif insertions sharing similarity with HIV-1 proteins could provide an enhanced affinity towards host cell receptors and increase the range of host cells of 2019-nCoV. This study implies that 2019-nCoV might be generated by gaining gene fragments from the HIV-1 genome.
Current report conducted careful examination of the sequences of 2019-nCoV, other CoV viruses and HIV-1 as well as GenBank database. Our results demonstrated no evidence that the sequences of these four inserts are HIV-1 specific or the 2019-nCoV viruses obtain these insertions from HIV-1. First, the results of blast search of these motifs against GenBank shows that the top 100 identical or highly homologous hits are all from host genes of mammalian, insects, bacterial and others. There are only a few hits on coronaviruses, but none of them are HIV-1 related. Blast against viral sequence database also showed these insertion sequences widely exist in all kinds of viruses from bacteriophage, influenza, to giant eukaryotic viruses (https://www.hiv.lanl.gov/components/sequence/HIV/search/search.html) yielded similar results. Sequences that completely match the insertion 3 and 4 sequences were not found in any HIV-1 sequences. This clearly shows that these insertioin sequences are widely present in living organisms including viruses, but not HIV-1 specific. All these regions in HIV-1 envelope glycoprotein are highly variable with many large insertions and deletions, indicating that they are not essential for biological functions of HIV-1 envelope glycoprotein. The detection of completely matched sequences of 1 and 2 insertions in only a few HIV-1 strains demonstrated that four insertions are very rare or not present among tens of thousands of natural HIV-1 sequences. This also explains why four insertion homolog sequences could only be independently found in different HIV-1 genomes [8]. Because of their poor identities to and rareness in the HIV-1 sequences, HIV-1 could not be the source for those insertion sequences in the 2019-nCoV genome.
Second, these insertions are present not only in 2019-nCoV viruses but also in three betaCoV sequences from bats: two (ZC45 and ZXC21) from Zhejiang deposited in GenBank in 2018 and RaTG13 from Yunnan obtained in 2013 [8]. The RaTG13 is much more similar to 2019-nCoV than both ZC45 and ZXC21 (Figure 1A). The similarity of the spike protein between RaTG13 and 2019-nCoV is 97.7%. In the RaTG13 genome, two inserts are identical (HKNNKS and RSYLTPGDSSSG) to those in 2019-nCoV, one has one T → I substitution (TNGIKR), and the fourth one misses the C-terminal 4 amino acids (QTNS----) (Figure 1B). ZC45 and ZXC21 are more divergent from 2019-nCoV than RaTG13, but both also contain similar insertions at three insertion sites, except insertion 4 (Figure 1B). Furthermore, many other CoV viruses have similar insertions but with different sequences at the insertion 1 position. These results clearly show that three out of four of these inserts naturally exist in three bat CoV viruses before 2019-nCoV was identified. This undoubtedly refutes the possibility that 2019-nCoV is generated through obtaining gene fragments from the HIV-1 genome. Instead, it is much more likely that 2019-nCoV originated from RaTG13-like CoV viruses.
Figure 1. Sequence and structure analysis of 2019-nCoV and bat coronaviruses. (A) Phylogenetic tree analysis of the spike gene sequences. (B) Sequence alignment of suspected insertion sites between the 2019-nCoV and bat coronavirus sequences. The deletions in the alignment are shown as dashes. The numbers of insertions are indicated at the top of the alignment. (C) Structure comparison of the four insertions in the CoV spike protein and HIV-1 gp120. 2019-nCoV structure was modelled using I-TASSER server with default parameters. Only relevant domains with residues 1 to 708 (exclude residues from 305 to 603) were presented as ribbon diagram. The four insertions were labelled and coloured in red, blue, green and magenta, respectively. HIV-1 gp120 structure (PDB 1GC1) is presented as ribbon diagram. V4, V5, V1/V2 and LE loops were labelled and coloured in red, blue, green, and black, respectively.
Display full size
Third, insertions 1 and 2 in 2019-nCoV have 6-AA motifs identical to those in V4 and V5 of certain HIV-1 gp120 isolates, which are structurally close to each other but separated by a LE loop (Figure 1C) [9]. However, insertion 3 located between insertions 1 and 2 in 2019-nCoV has sequences similar (with deletions) to those in the V1 region of HIV-1 gp120. V1 is far away from V4 and V5 on the opposite side of gp120, which should not interact with V4/V5 in gp120 (Figure 1C) but is now inserted between V4 and V5 in the modelled the 2019-nCoV spike protein structure [10]. Insertion 4 was found in Gag protein of HIV-1 that is not associated with viral entry. This insertion is located too far to be considered to form the same structural unit with the other three insertions in the 2019-nCoV spike protein (Figure 1C). We do not see any selection benefit or rationale for 2019-nCoV to obtain and mix structurally unrelated parts of HIV-1 to generate a unique structure for its enhanced receptor binding as indicated by the authors [8].
How the three bat CoV viruses obtain those inserts remains unknown. For any virus to obtain additional insert sequences from other organisms, it requires that it has direct interactions with other organisms, most likely through homologous or non-homologous recombination [11]. For bat CoV viruses to gain the gene fragments from HIV-1, it will require both viruses to co-infect the same cells. Because the host cells for bat CoV viruses and HIV-1 are different, the chance for both to exchange genetic materials is negligible. On the contrary, these motifs are widely present in various mammalian cells and so it will be more likely for bat CoV viruses to gain those motifs from the genomes of their infected cells if recombination indeed occurs. However, extensive studies of more CoV viruses in wild and domestic animals are warranted to address this question.
Identification of the origins of these inserted sequences in three bat CoV viruses and the new epidemic 2019-nCoV strain will be important for us to understand how CoV viruses jump from animals to humans and adapt in the latter. Current data showed that RaTG13 is most closely related to 2019-nCoV [7]. However, the genetic difference between them is too high for RaTG13 to serve as the immediate ancestor of 2019-nCoV. Other viruses that are more closely related to 2019-nCoV in intermediate animals like civet for SARS and camel for MERS [3,12] are remained to be identified. More studies are necessary to identify the real source of 2019-nCoV. This may take a long time to identify the origin of 2019-nCoV by screening a large number of wild and domestic animals. In any case, reducing or eliminating direct contacts with wild animals will be critical to control the new epidemic infection diseases in the future.
The advances in bioinformatics analysis tools are widely used to easily and rapidly analyse newly obtained sequences. However, great care is required for comprehensive and thorough analysis to fully understand the real biological implications of the new genomic information. Biased, partial and incorrect analysis can dangerously lead to conclusions that fuel conspiracies and harm the process of true scientific discoveries and the effort to control the damage to public health.
Last edited:
Copy Ninja
Superstar
and it's being treated with HIV/AIDS retrovirals.
I should also be clear - antiretrovirals are often broad-spectrum drugs that can be used effectively against many RNA-based viruses. Just like an antibiotic like penicillin isn't effective as just one bacterial disease, many of the antiretrovirals used to treat AIDS patients can also be used against other RNA-based viral diseases.
For example, I clicked on one HIV antiretroviral drug at random (Raltegravir), and found immediately that it also appears to be effective against herpes.
newworldafro
DeeperThanRapBiggerThanHH
It was retracted because it was bullshyt. The paper hadn't been reviewed by anyone and within hours more careful scientists were poking a thousand holes in the claim. Some easy examples of that are in the Twitter thread here:
You don't have to follow the mainstream media on this if you don't want to. There are tens of thousands of microbiologists and infectious disease specialists all over the world who have the same access to that article that you do. I've been part of that world before - trust me, they aren't a bunch of unthinking establishment lackeys and they aren't obeying instructions from a hive mind. If there is any legitimacy to the paper or any real connection between COVID-19 and HIV, you will see plenty of virologists talking about it no matter what the mainstream media says.
Here's a more formal comment declaring no contribution from HIV-1 in COVID-19:
https://www.tandfonline.com/doi/full/10.1080/22221751.2020.1727299
HIV-1 did not contribute to the 2019-nCoV genome
Chuan Xiao,Xiaojun Li,Shuying Liu,Yongming Sang,Shou-Jiang Gao &Feng Gao
Pages 378-381 | Received 04 Feb 2020, Accepted 04 Feb 2020, Published online: 14 Feb 2020
When a new pathogen that causes a global epidemic in humans, one key question is where it comes from. This is especially important for a zoonotic infectious disease that jumps from animals to humans. Knowing the origin of such a pathogen is critical to develop means to block further transmission and to develop vaccines. Discovery of the origin of a newly human pathogen is a sophisticated process that requires extensive and vigorous scientific validations and generally takes many years, such as the cases for HIV-1 [1], SARS [2] and MERS [3]. Unfortunately, before the natural sources of new pathogens are clearly defined, conspiracy theories that the new pathogens are man-made often surface as the source. However, in all cases, such theories have been debunked in history.
Infection from an emerging pathogenic coronavirus was first reported in December 2019 in China. It has now affected over 42,000 people and caused over 1,000 deaths in 25 countries (https://2019ncov.Chinacdc.Cn/2019-Ncov). The complete genome of this new virus was quickly sequenced and made public on January 12, only about 2 weeks after the disease was first observed [4]. It was named as 2019-nCoV the following day by the World Health Organization (WHO). Phylogenetic analysis shows that 2019-nCoV is a new member of coronaviruses that infect humans. It is genetically homogenous but distinct from coronaviruses that cause SARS and MERS [5,6]. However, it shares a high level of genetic similarity (96.3%) with a bat coronavirus RaTG13 which was obtained from bat in Yunnan in 2013, suggesting that RaTG13-like viruses are most likely the reservoir, but not the immediate sources of the current 2019-nCoV viruses [7].
Lack of the definite origin of 2019-nCoV has led to speculation that 2019-nCoV might be derived from genetic manipulation or even for the purpose of use as a bioweapon. This notion has been fully debunked in the media. A recent informally presented report, however, showed that 2019-nCoV had four insertions in the spike glycoprotein gene that is critical for the virus to enter the target cells when compared to other coronaviruses [8]. It was claimed that these inserts were either identical or similar to the motifs in the highly variable (V) regions (V1, V4 and V5) in the envelope glycoprotein or in the Gag protein of some unique HIV-1 strains from three different countries (Thailand, Kenya and India). Together with the structure modelling analysis, the authors speculated that these motif insertions sharing similarity with HIV-1 proteins could provide an enhanced affinity towards host cell receptors and increase the range of host cells of 2019-nCoV. This study implies that 2019-nCoV might be generated by gaining gene fragments from the HIV-1 genome.
Current report conducted careful examination of the sequences of 2019-nCoV, other CoV viruses and HIV-1 as well as GenBank database. Our results demonstrated no evidence that the sequences of these four inserts are HIV-1 specific or the 2019-nCoV viruses obtain these insertions from HIV-1. First, the results of blast search of these motifs against GenBank shows that the top 100 identical or highly homologous hits are all from host genes of mammalian, insects, bacterial and others. There are only a few hits on coronaviruses, but none of them are HIV-1 related. Blast against viral sequence database also showed these insertion sequences widely exist in all kinds of viruses from bacteriophage, influenza, to giant eukaryotic viruses (https://www.hiv.lanl.gov/components/sequence/HIV/search/search.html) yielded similar results. Sequences that completely match the insertion 3 and 4 sequences were not found in any HIV-1 sequences. This clearly shows that these insertioin sequences are widely present in living organisms including viruses, but not HIV-1 specific. All these regions in HIV-1 envelope glycoprotein are highly variable with many large insertions and deletions, indicating that they are not essential for biological functions of HIV-1 envelope glycoprotein. The detection of completely matched sequences of 1 and 2 insertions in only a few HIV-1 strains demonstrated that four insertions are very rare or not present among tens of thousands of natural HIV-1 sequences. This also explains why four insertion homolog sequences could only be independently found in different HIV-1 genomes [8]. Because of their poor identities to and rareness in the HIV-1 sequences, HIV-1 could not be the source for those insertion sequences in the 2019-nCoV genome.
Second, these insertions are present not only in 2019-nCoV viruses but also in three betaCoV sequences from bats: two (ZC45 and ZXC21) from Zhejiang deposited in GenBank in 2018 and RaTG13 from Yunnan obtained in 2013 [8]. The RaTG13 is much more similar to 2019-nCoV than both ZC45 and ZXC21 (Figure 1A). The similarity of the spike protein between RaTG13 and 2019-nCoV is 97.7%. In the RaTG13 genome, two inserts are identical (HKNNKS and RSYLTPGDSSSG) to those in 2019-nCoV, one has one T → I substitution (TNGIKR), and the fourth one misses the C-terminal 4 amino acids (QTNS----) (Figure 1B). ZC45 and ZXC21 are more divergent from 2019-nCoV than RaTG13, but both also contain similar insertions at three insertion sites, except insertion 4 (Figure 1B). Furthermore, many other CoV viruses have similar insertions but with different sequences at the insertion 1 position. These results clearly show that three out of four of these inserts naturally exist in three bat CoV viruses before 2019-nCoV was identified. This undoubtedly refutes the possibility that 2019-nCoV is generated through obtaining gene fragments from the HIV-1 genome. Instead, it is much more likely that 2019-nCoV originated from RaTG13-like CoV viruses.
Figure 1. Sequence and structure analysis of 2019-nCoV and bat coronaviruses. (A) Phylogenetic tree analysis of the spike gene sequences. (B) Sequence alignment of suspected insertion sites between the 2019-nCoV and bat coronavirus sequences. The deletions in the alignment are shown as dashes. The numbers of insertions are indicated at the top of the alignment. (C) Structure comparison of the four insertions in the CoV spike protein and HIV-1 gp120. 2019-nCoV structure was modelled using I-TASSER server with default parameters. Only relevant domains with residues 1 to 708 (exclude residues from 305 to 603) were presented as ribbon diagram. The four insertions were labelled and coloured in red, blue, green and magenta, respectively. HIV-1 gp120 structure (PDB 1GC1) is presented as ribbon diagram. V4, V5, V1/V2 and LE loops were labelled and coloured in red, blue, green, and black, respectively.
Display full size
Third, insertions 1 and 2 in 2019-nCoV have 6-AA motifs identical to those in V4 and V5 of certain HIV-1 gp120 isolates, which are structurally close to each other but separated by a LE loop (Figure 1C) [9]. However, insertion 3 located between insertions 1 and 2 in 2019-nCoV has sequences similar (with deletions) to those in the V1 region of HIV-1 gp120. V1 is far away from V4 and V5 on the opposite side of gp120, which should not interact with V4/V5 in gp120 (Figure 1C) but is now inserted between V4 and V5 in the modelled the 2019-nCoV spike protein structure [10]. Insertion 4 was found in Gag protein of HIV-1 that is not associated with viral entry. This insertion is located too far to be considered to form the same structural unit with the other three insertions in the 2019-nCoV spike protein (Figure 1C). We do not see any selection benefit or rationale for 2019-nCoV to obtain and mix structurally unrelated parts of HIV-1 to generate a unique structure for its enhanced receptor binding as indicated by the authors [8].
How the three bat CoV viruses obtain those inserts remains unknown. For any virus to obtain additional insert sequences from other organisms, it requires that it has direct interactions with other organisms, most likely through homologous or non-homologous recombination [11]. For bat CoV viruses to gain the gene fragments from HIV-1, it will require both viruses to co-infect the same cells. Because the host cells for bat CoV viruses and HIV-1 are different, the chance for both to exchange genetic materials is negligible. On the contrary, these motifs are widely present in various mammalian cells and so it will be more likely for bat CoV viruses to gain those motifs from the genomes of their infected cells if recombination indeed occurs. However, extensive studies of more CoV viruses in wild and domestic animals are warranted to address this question.
Identification of the origins of these inserted sequences in three bat CoV viruses and the new epidemic 2019-nCoV strain will be important for us to understand how CoV viruses jump from animals to humans and adapt in the latter. Current data showed that RaTG13 is most closely related to 2019-nCoV [7]. However, the genetic difference between them is too high for RaTG13 to serve as the immediate ancestor of 2019-nCoV. Other viruses that are more closely related to 2019-nCoV in intermediate animals like civet for SARS and camel for MERS [3,12] are remained to be identified. More studies are necessary to identify the real source of 2019-nCoV. This may take a long time to identify the origin of 2019-nCoV by screening a large number of wild and domestic animals. In any case, reducing or eliminating direct contacts with wild animals will be critical to control the new epidemic infection diseases in the future.
The advances in bioinformatics analysis tools are widely used to easily and rapidly analyse newly obtained sequences. However, great care is required for comprehensive and thorough analysis to fully understand the real biological implications of the new genomic information. Biased, partial and incorrect analysis can dangerously lead to conclusions that fuel conspiracies and harm the process of true scientific discoveries and the effort to control the damage to public health.
So you how to explain Chinese scientist saying it has HIV/AIDS components and the part about it being treated with HIV/AIDS meds. Ok.
Ok. Indian scientist needed more scientific scrutiny, but now Chinese scientists are supporting the Indians' "voluntary retracted" original conclusion.
I agree with you it's important to have scientific analysis that is checked and re-checked, so proper information is getting out there.
That's why we having this open dialogue and I hope it stays this way.